CTNNB1 Treatment Strategies
We are fighting for treatment solutions that could help all affected patients. While severe phenotype-associated mutations may require gene therapy (strategies 1 and 3), milder phenotype-associated mutations could be treated with less invasive treatments options (strategy 2).
Strategy 1: Gene Replacement Therapy
Gene replacement therapy (GRT) is a one-time treatment that delivers a healthy copy of the gene to the cells with viral vectors. The majority of patients with a CTNNB1 syndrome have one mutated gene that causes loss-of-function of the protein called beta-catenin.
Gene replacement therapy is a type of therapy where a functional copy of CTNNB1 is delivered to a patient’s cells through adeno-associated virus (AAV) delivery. The intended administration route is intrathecal administration.
Gene Therapy, in general, is an umbrella term for delivering targeted genetic material to replace, modify or suppress gene expression in patients affected with genetic diseases. It comprises different technologies (antisense, virus-mediated gene replacement, and other gene-directed therapies) that have been very recently developed with a potential to cure severe genetic diseases.
Technologies are progressing rapidly, with press releases announcing big breakthroughs every month. Based on the report from January 2021, there are currently 284 gene therapy programs available for 130 different disease conditions (www.bluematrix.com).
Gene therapies have made a huge progress since the US approval of Luxturna (Spark/Roche) and Zolgensma (AveXis/Novartis) in 2019. Both of these programs have demonstrated that gene therapy can be successfully delivered to different tissues and that they can be very efficient.
Luxturna was the first gene therapy to treat an inherited retinal disease for children and adults with vision loss caused by a mutation in the RPE65 gene. A one-time treatment designed to provide a working copy of the not working gene restores vision and improves sight (www.novartis.com)
A one-time treatment designed for children with SMA, has also shown significant unimaginable benefits. Presymptomatic babies with SMA treated with Zolgesma soon after birth had no delays in achieving milestones. Normally, these babies would die before their second birthday (www.novartis.com). The growing number of children who are now, thanks to Zolgensma, meeting their motor milestones in a timely manner, demonstrates the promise of gene therapy.
Strategy 2: RNA-based Therapies
CTNNB1 Syndrome is an autosomal dominant condition. While one gene is mutated, the other one is healthy. The RNA therapies would enhance the wild type β-catenin from the healthy CTNNB1 allele. Inhibition of β-catenin degradation is currently explored by a) Skipping exon3 that encodes the regulatory region, b) Editing of β-catenin mRNA to mutate key phosphorylation residues that govern degradation, c) Downregulation of proteins involved in the β-catenin destruction complex.
Strategy 3: DNA-modification techniques
A “search and replace” gene editing method that can correct mutations in a precise way.
Our researchers will work on the development of prime editing variation for genome editing for delivery via viral or nonviral (RNP, RNA) delivery. Currently, this is tested on a reporter and if successful on relevant cells harboring the specific CTNNB1 mutation.
Our gene therapy program – URBAGEN
URBAGEN is being developed to be a single-dose intracerebroventricular injection of non-replicating, single-stranded AAV9 vector for loss-of-function variants in the CTNNB1 syndrome.
It contains the human CTNNB1 gene, which encodes for the beta-catenin protein, under the control of the cytomegalovirus-enhanced chicken-beta-actin hybrid (CBh) promoter.
The size of the packaged single-stranded vector genome is ~4.7 kb, which is ideal for the ctnnb1 gene encoding β-catenin, as it ensures effective transport without degradation, enhancing gene therapy efficacy.
Within our project, more than a dozen therapeutic CTNNB1 gene therapy variants with the CBh promoter have been designed, and some of them have passed early in silico evaluation and were cloned for in vitro and in vivo evaluation.
Initial testings were performed in iNPCs in order to triage the best ones for in vivo assessment. Within each candidate researchers have systematically evaluated regulatory elements in both the coding and non-coding components to ensure both therapeutic efficacy and safety profiles of any therapeutic lead.
These constructs were individually packaged into AAV vectors, and the vectors were used to transduce patient-derived neuro-progenitor cells and cortical brain organoids. We found that only 1 of our AAV-CTNNB1 constructs was able to restore β-catenin expression levels and function in both preclinical models.
The AAV-CTNNB1 construct was tested for efficacy in a disease mouse model from Jackson Laboratory. Three doses of AAV9-Construct4 were given via ICV route to a 28 days old mice (corresponds to 6-12 years old human) , and behavioral tests showed phenotype improvements. High doses enhanced anxiety, in male activity and motor skills and mid doses improved gait in both sexes. Brain analysis showed increased hCTNNB1 expression correlating with dosage. Further studies in wild type mice monitored behavior, physical health, and organ collection. (Information regarding the safety of AAV gene therapy)
In November 2023, we have initiated manufacturing process with our European partner Viralgen VC. Our construct has passed 2l development and the comparability with the research-grade vector was acceptable. At this moment, we are developing 50l toxicology batch and will start with toxicology testings in October/November 2024. We expect to start with the 250l clinical batch manufacturing early 2025.
The proprietary technology underlying Urbagen is protected by licensed patent rights (application number 2023903490), filed by Children’s Medical Research Institute (CMRI) on 31st of October 2023, based on the work of Dr Leszek Lisowski. The CTNNB1 Foundation has obtained from CMRI an exclusive, worldwide license for the development and commercialization of Urbagen.
This license agreement underscores the commitment to address the unmet need within the CTNNB1 syndrome community and creates a clear pathway for bringing this breakthrough therapy to patients worldwide.
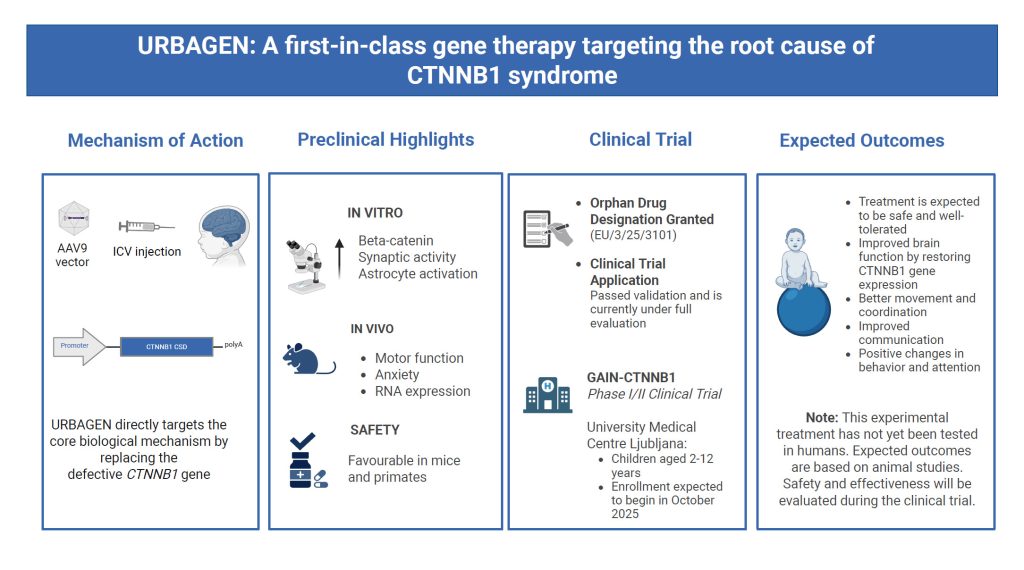
Our gene therapy, URBAGEN, has officially been granted an Orphan Drug Designation for the treatment of CTNNB1 syndrome by the European Medicines Agency (EMA) on 18 July 2025. This is a major step forward on our path toward clinical trials and, one day, approved treatment.
What does this mean?
CTNNB1 syndrome is an ultra-rare genetic condition, affecting only a small number of individuals worldwide. Because there are so few patients, developing a treatment is extremely expensive and often not prioritized by big pharmaceutical companies. That’s where Orphan Drug Designation comes in. This special status is granted to therapies for rare diseases and comes with support and incentives from regulators – like help with the development process, reduced fees, and future market protection. It makes it more possible (and realistic) to bring life- changing treatments like URBAGEN to patients who need them most.
The medicinal product, formally named “Adeno-associated virus vector serotype 9 containing the human CTNNB1 gene”, is now officially listed in the Community Register of Orphan Medicinal Products under the number EU/3/25/3101. This recognition from European regulators strongly affirms the scientific foundation of our work and highlights their acknowledgment of both the urgency and potential of our therapy.